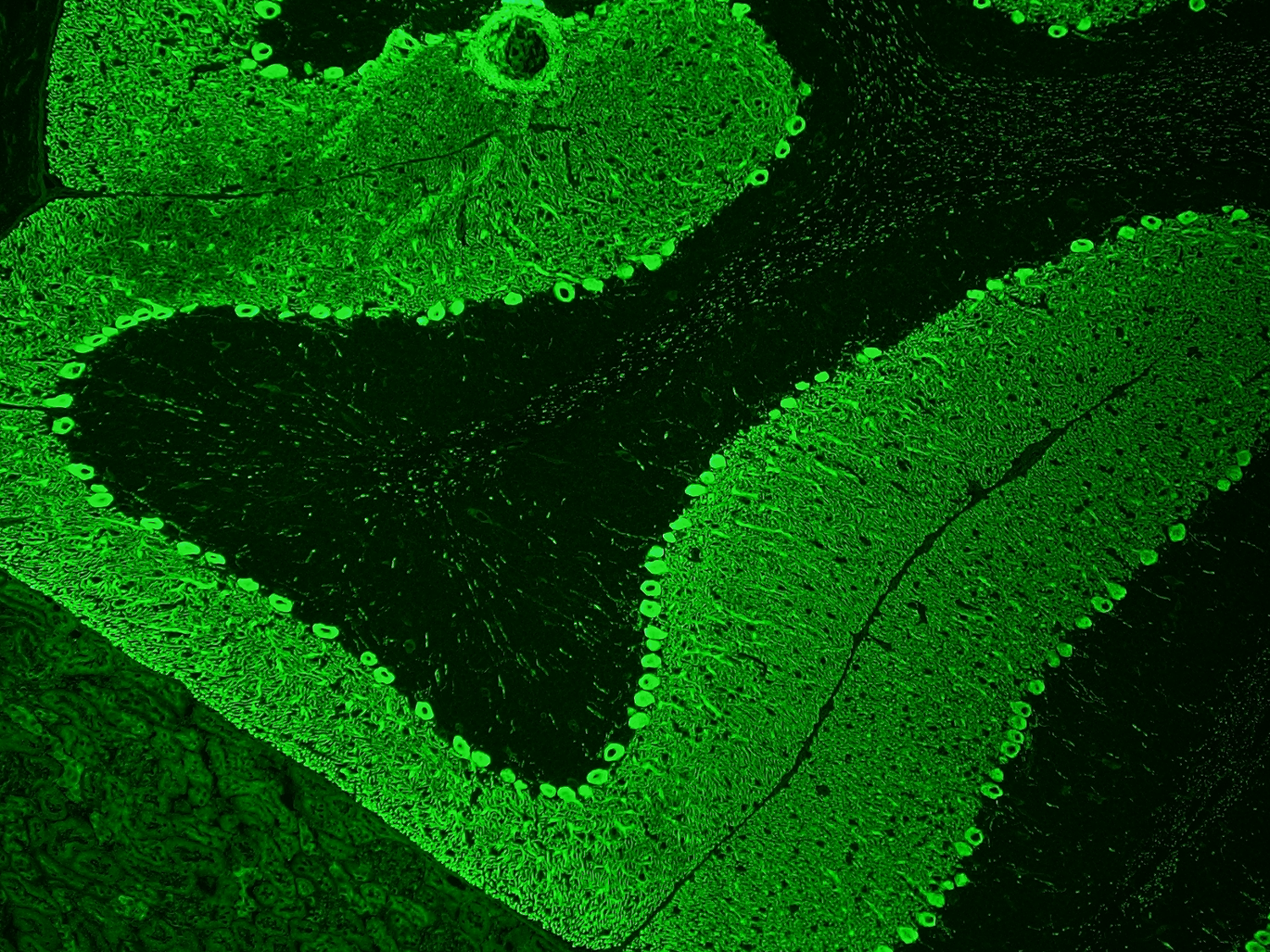
")
Dr. Avi Gadoth , Lab PI
Email: avig@tlvmc.gov.il
Phone: +972-3-6973772

Valeria Briskin PhD, Lab manager
Phone: +972-3-6974767
Email: valeriabr@tlvmc.gov.il








Email: avig@tlvmc.gov.il
Phone: +972-3-6973772
Phone: +972-3-6974767
Email: valeriabr@tlvmc.gov.il